



A Comprehensive Guide to De Novo Genome Assembly

Posted by kikoetgarcia
from the Health category at
28 Jun 2023 09:40:54 am.
Share this page: 












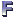






Types of Reads
To accomplish genome assembly using next-generation sequencing platforms, computer programs typically utilize single reads and paired reads. The length of these "reads" varies between 20 and 1000 base pairs (bp) depending on the sequencing platform employed. Single reads are short sequenced fragments that can be combined based on overlapping regions to form a continuous sequence called a "contig." On the other hand, paired reads are approximately the same length as single reads but originate from opposite ends of DNA fragments. Paired reads are preferred over single reads since they aid in linking contigs into "scaffolds" and provide insights into the size of repetitive regions.
However, challenges such as repetitive sequences, variants, missing data, and errors sometimes limit the efficiency and accuracy of genome assembly. Long-read technologies have emerged to address these limitations by spanning stretches of repetitive regions and generating a continuous reconstruction of the genome. Currently, two dominant methods in this new generation are single molecule real-time (SMRT) sequencing, championed by Pacific Biosciences (PacBio), and nanopore sequencing, championed by Oxford Nanopore Technologies. SMARTdenovo, an assembly pipeline utilizing long reads, has been demonstrated to produce reasonably high-quality assemblies from both MinION and SMRT reads.
Illumina Genome Assembly
To illustrate the workflow of genome assembly with next-generation sequencing (NGS) data, we will employ Illumina genome assembly as an example. Illumina sequencing is one of the most commonly used approaches in genomics studies.
Assessing Read Quality
Before proceeding with genome assembly, it is crucial to evaluate the quality of the sequence data to avoid drawing erroneous conclusions. The reads can be stored in formats such as Fasta, FastQ, SAM, and BAM. FastQ is the most commonly used read file format, particularly in data produced by the Illumina sequencing pipeline. Besides read types, other factors such as the number of reads, GC content, and contamination also need to be considered.
Base calling accuracy, which assesses the probability of a given base being called incorrectly, is commonly determined using Phred quality scores (Q scores). FastQC is a widely used tool for quality control of raw data. Its main outputs include read length, quality encoding type, %GC content, total number of reads, presence of highly recurring k-mers, presence of numerous N's in reads, and dips in quality at the beginning, middle, or end of the reads.
Pre-processing Raw Data
Once the quality of the sequence data is assessed, various tools for quality trimming are available in platforms like Galaxy or through command-line interfaces, such as Trimmomatic. Trimmomatic can handle read pairing when paired reads are available. It performs multiple trimming functions sequentially, including:
Adapter trimming: Removing adapters, barcodes, and other contaminants.
Sliding window trimming: Evaluating average quality and trimming accordingly.
Bases quality trimming: Trimming low-quality trailing and leading bases.
Minimum read length: Ensuring that the reads, after all trimming steps, exceed the minimum read length. If not, the reads are discarded.
Another tool, PRINSEQ, serves a similar purpose of quality trimming raw data.
De Novo Genome Assembly
The next step involves assembling the quality-trimmed reads into draft contigs. The recommended assembly software for this stage is Velvet Optimiser, which utilizes the Velvet Assembler. The Velvet Assembler is specifically designed for short reads in Illumina style and employs the de Bruijn graph approach. Both Velvet Assembler and Velvet Optimiser can handle multiple read files (e.g., SAM, BAM, FastQ, and Fasta) and types (e.g., single-ended, paired-end, and mate pair). The quality of contigs assembled by Velvet largely depends on parameter settings, with critical parameters including hash size, expected coverage, and coverage cutoff. Alternative de novo assemblers include Spades, SOAP-denovo, MIRA, and ALLPATHS.
Assembly Polishing
After completing the aforementioned steps, the draft contigs obtained may contain gaps or regions represented by 'N's, and some of them may be misassembled. To refine the assembly, misassembly checking tools and assembly metric tools such as QUAST, InGAP-SV, and Mauve assembly metrics can be employed.
To achieve a finished genome, additional data from different sources or alternative tools can be utilized. Genome finishing tools encompass semi-automated gap fillers (e.g., Gap filler), genome visualizers and editors (e.g., Artemis, IGV, Geneious, CLC BioWorkbench), and annotation tools (e.g., Prokka, RAST, and JCVI Annotation Service).
0 Comments